research interests
novel molecular technologies to study chromatin & transcription
Our lab develops high-throughput molecular tools to study how mammalian genes are regulated in the context of chromatin – the nucleopotein complex responsible for packaging, protecting, & interpreting eukaryotic DNA. Work in our lab has been propelled by advances in single-molecule, long-read nucleic acid sequencing, which offers unique advantages over prevailing “short-read” high-throughput DNA sequencing. For instance, we developed the single-molecule adenine methylated oligonucleosome sequencing assay (SAMOSA), one of the first assays to combine methylase footprinting of chromatin with long-read sequencing on the Pacific Biosciences (PacBio) instrument, to map protein-DNA interactions on individual DNA molecules at scale. We now apply SAMOSA and offshoots of the approach to build a quantitative & mechanistic understanding of how cis and trans regulators interact with chromatin. While the lab’s research mainly focuses on basic chromatin biology, we are also generally interested in developing new methods that use long-read sequencing. For instance, we developed SMRT-Tag – a transposase-based approach that enables native PacBio DNA sequencing of very small amounts of input DNA. We have also worked to improve single-molecule RNA sequencing methods; working with Hani Goodarzi’s group, we’ve begun experiments to better understand how chromatin instructs the alternative splicing of mRNA.
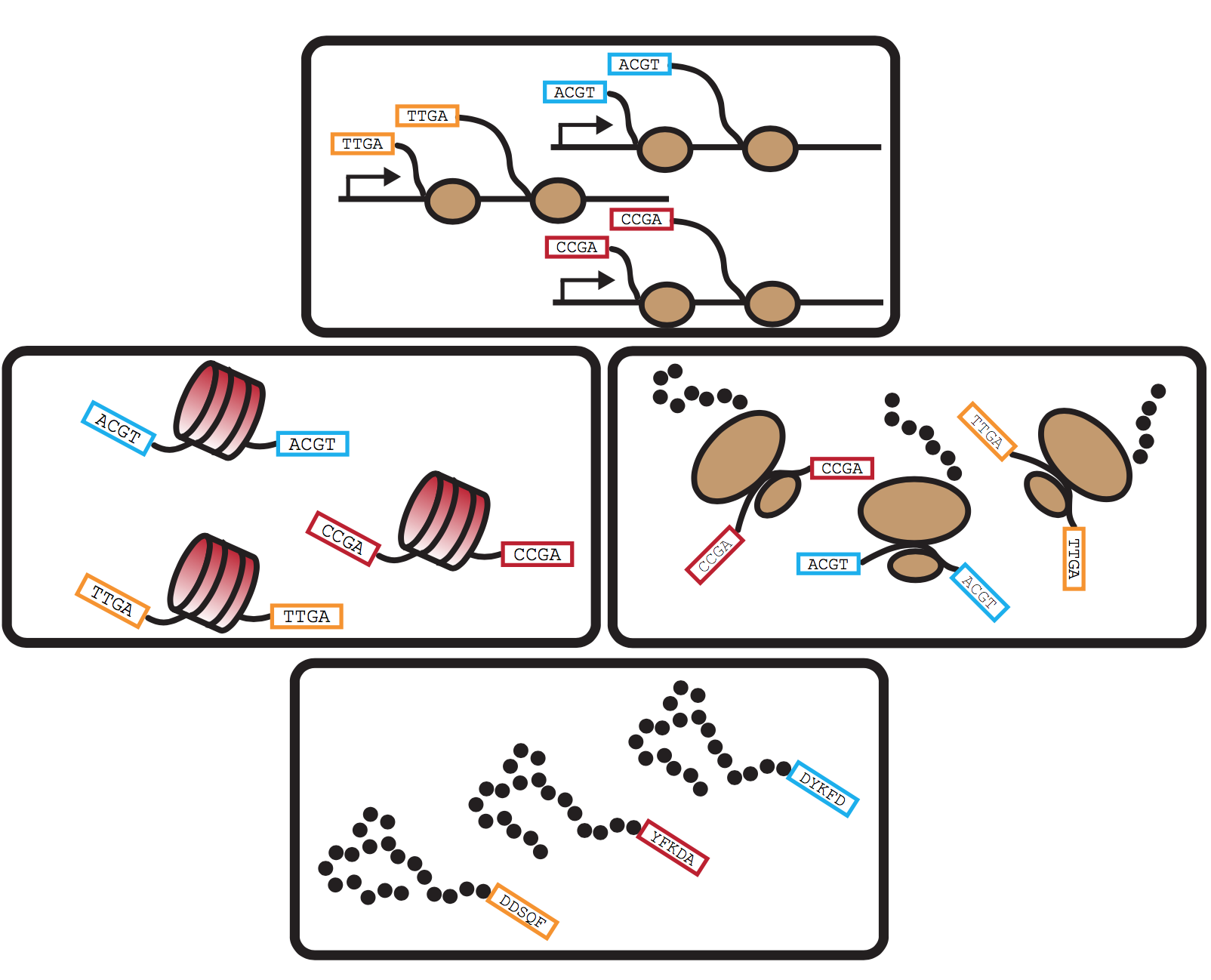
in vivo mapping & quantitative in vitro reconstitution of chromatin-based regulation
Genomic technologies provide invaluable descriptive views of gene regulatory processes, but often fall short of providing causal mechanistic insight. To address this limitation, we’ve worked closely with Geeta Narlikar’s lab, to combine SAMOSA footprinting with precise biochemical reconstitution of chromatin fibers in vitro. As proof-of-concept of this approach, we examined the activity of the human ISWI chromatin remodeling complex on chromatin reconstituted on mammalian genomic sequence, demonstrating through a combination of in vitro and in vivo single-molecule experiments that these remodelers sense nucleosome density to program the nucleosome spacing on individual DNA templates. We are actively applying this approach to quantitatively reconstitute more complex chromatin “computation,” with the goal of complementing the troves of in vivo genomic mapping data currently available.
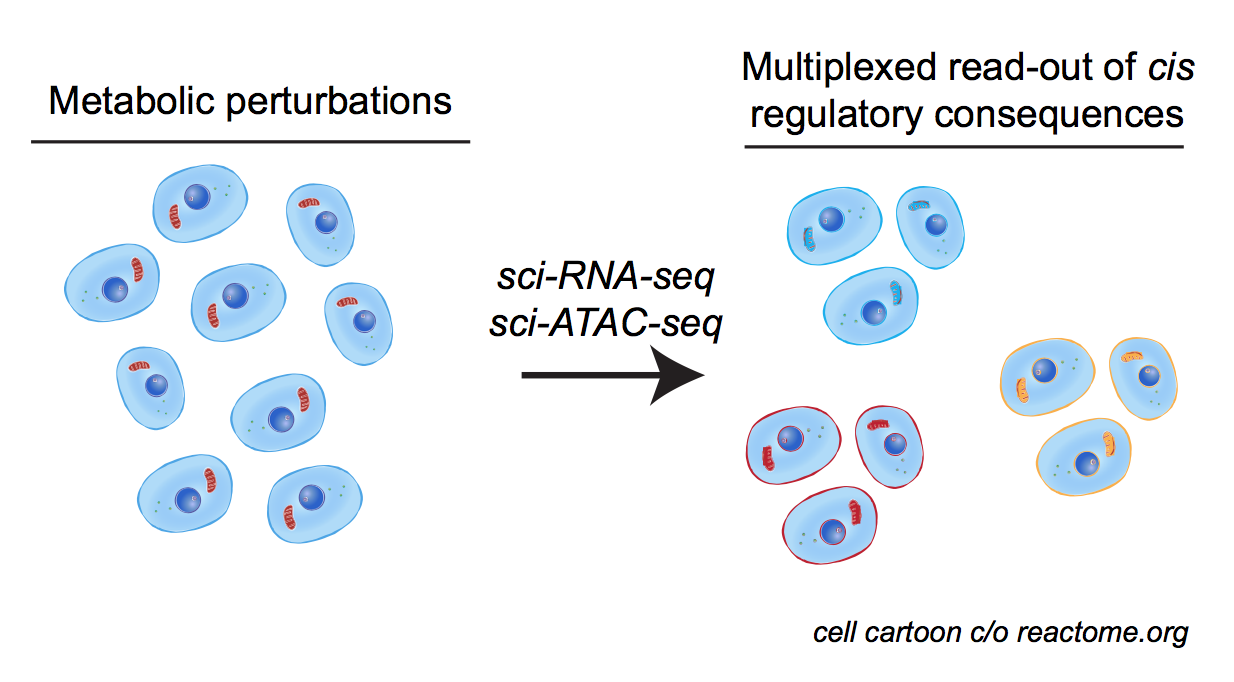
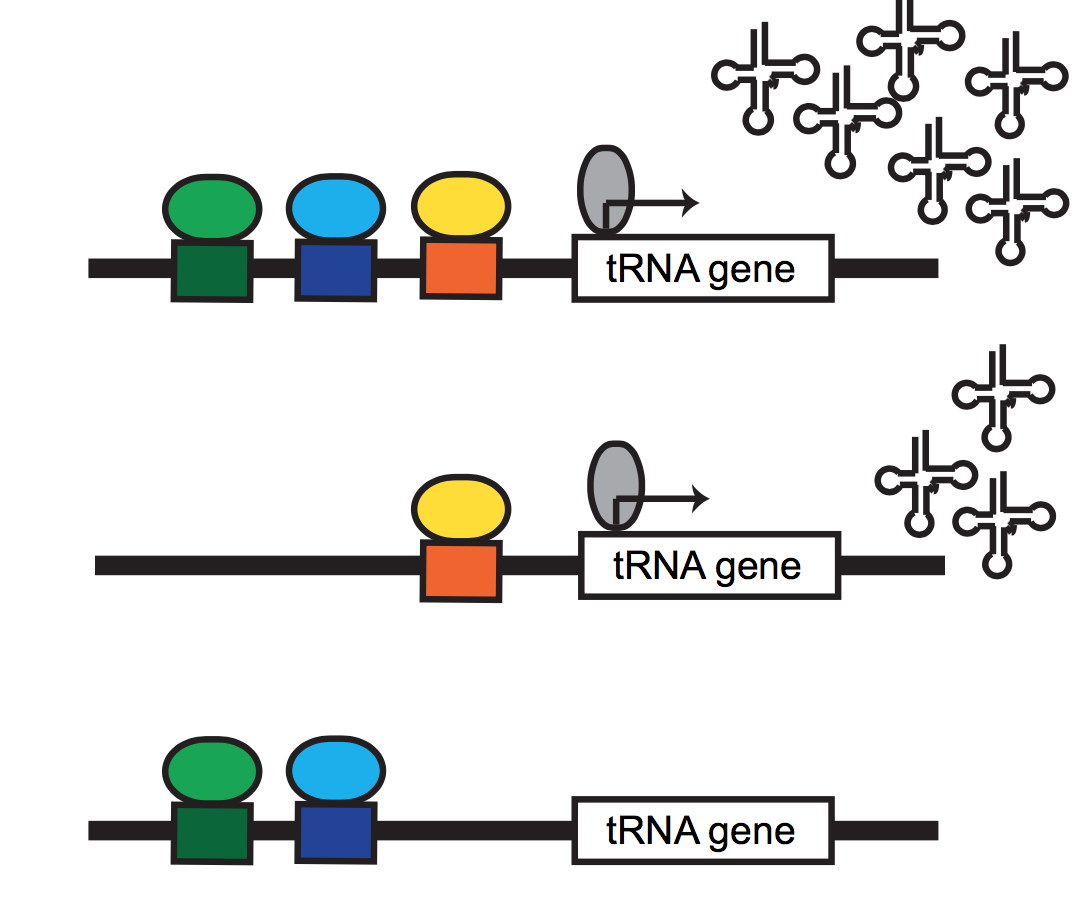
physiological chromatin computation in health & disease
Cellular & organismal functions in development & disease rely on the proper integration of extracellular signals into gene regulatory responses. We are broadly interested in understanding how cellular stressors (e.g. metabolic perturbation, hormone signaling, etc.) are sensed by the dynamic epigenome, and associated memory for such responses. This broad interest has taken us in a variety of directions: we have collaborated with Bob Eisenman’s lab at the Fred Hutchinson Cancer Research Center, to apply our tools to diverse in vitro and in vivo models of MYC network dysregulation; with Hani Goodarzi and many other members of UCSF’s Helen Diller Cancer Center, we’ve begun charting the chromatin structure associated with tumour indolence & metastasis; also with the Goodarzi group, we’ve begun dissecting the regulatory elements necessary for RNA Polymerase III transcription in mouse and human, in hopes of defining new metabolically-responsive cis & trans regulatory elements.